


What are foundation models for? Lessons from synbio
What can other fields teach us about foundation models?

I’ve been taking a synthetic biology class1 and have been amazed at what machine learning can do in this field. A lot of tools qualify as foundation models, which has been the subject of many blog posts here in the context of neuroscience. I think synbio holds many lessons for the next generation of modeling for neuroscience. In this blog post, I will go through some of the tools available in synbio, and try to learn what neuroscience might look like in the (hopefully not too distant) future.
In the past, I’ve introduced foundation models for neuroscience with analogy to large-language models. Many different paradigms can be integrated into language models: anything that can be a token will eventually get tokenized. I’ve started to think this is not quite the right mental model for foundation models in neuroscience, putting too much emphasis on a single monolithic model and not enough on the ecosystem that surrounds it. Let’s unpack this.
Foundation models in synbio
In synthetic biology and computational biology more generally, proteins are a core unit of interest. Their sequence, physical shapes, activity, and interactions are of core interest. I’m sure many of you will be familiar with the Nobel-prize winning AlphaFold, which predicts protein structure from their sequence. These were built on decades of painstakingly reconstructing protein structure through techniques like cryo-EM and X-ray crystallography, aggregated in databases like PDB (protein database). But protein-folding is only application one of an ebullient slate of large-scale models. In addition to folding models, there are protein language models like ESM, which are trained to learn representations of residue sequences (that is, one token = one of 20 residues). There are genome language models like HyenaDNA that can reason over chunks of DNA with more than a million nucleotides; and there are multimodal models like Evo 2 that deal in DNA, RNA and proteins.
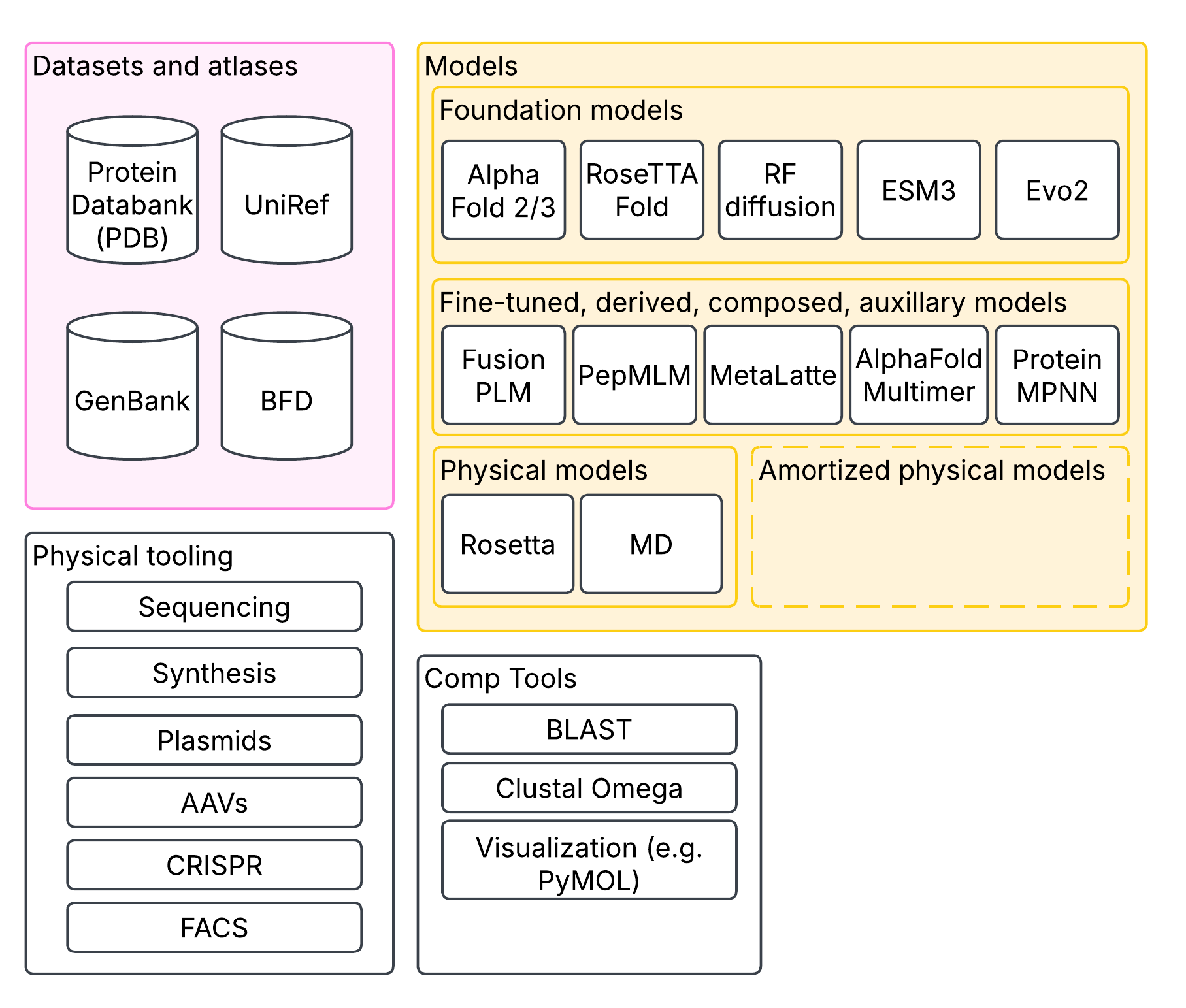
Some extensions can model multiple proteins interacting together, like AlphaFold Multimer; there are models that can design proteins from specifications, like RFDiffusion; there are inverse protein folding models that go from structure to sequence, like ProteinMPNN. Then there’s fine-tuned models, like the many models that build on top of ESM to predict protein activity across diverse domains. All of this is enabled by dozens of open tools to browse proteins and sequences, find similar sequences, and visualize structure; and databases of protein structures, sequences, measurements and predictions that have been built over decades. Molecular dynamics and other conventional physical modeling tools like Rosetta have not gone away either, forming an important complementary set of tools to machine learning-based models. Physical models can also be used to train machine learning models for amortized inference.
Importantly, we have the ability to act on this information! We can synthesize sequences, put them in plasmids to duplicate them, pack them in AAVs, edit cells with CRISPR, verify our edits through sequencing, etc. Put together, this vastly accelerates the work of the synthetic or computational biologist. None of these models are perfect; but put together, they remove tedious and lengthy physical steps of optimization with surrogate endpoints, in silico. Surrogate endpoints are no stranger to biology: we use mouse models of disease all the time. But surrogates can be biased, and often fail the translation step to humans; the best surrogates are both fast and accurate.
Consider this recent Science paper on EVOLVEPro, a system to optimize protein designs. Protein design is a combinatorial problem. There are 20^N peptides with N residues, which for 100 residue proteins is larger than the number of atoms in the universe; exhaustively searching the space is a no-go. Rational design based on The results are outlined in the abstract:
EVOLVEpro outperformed zero-shot methods in benchmarks across 12 deep mutational scanning datasets, including epitope binding, nucleic acid binding, and enzyme catalysis. We used EVOLVEpro to engineer six different proteins with diverse applications. We improved the binding affinity two monoclonal antibodies by up to 40-fold, the indel formation activity of a miniature CRISPR nuclease by fivefold, the insertion efficiency of a prime editor by twofold, the integration efficiency of a serine integrase by fourfold, and the transcription fidelity and mRNA quality of a T7 RNA polymerase by 100-fold. (emphasis mine)
That sounds impressive! What is this mysterious system that can improve existing proteins on some metrics by 1-2 orders of magnitude? It’s conceptually simple: a foundation model for proteins is fitted with a prediction head, which is fit on baseline mutations to predict some desirable metric. The predicted best proteins are synthesized and assayed; the model is re-fit; the new best candidates are synthesized; and the process continues for a few iterations.
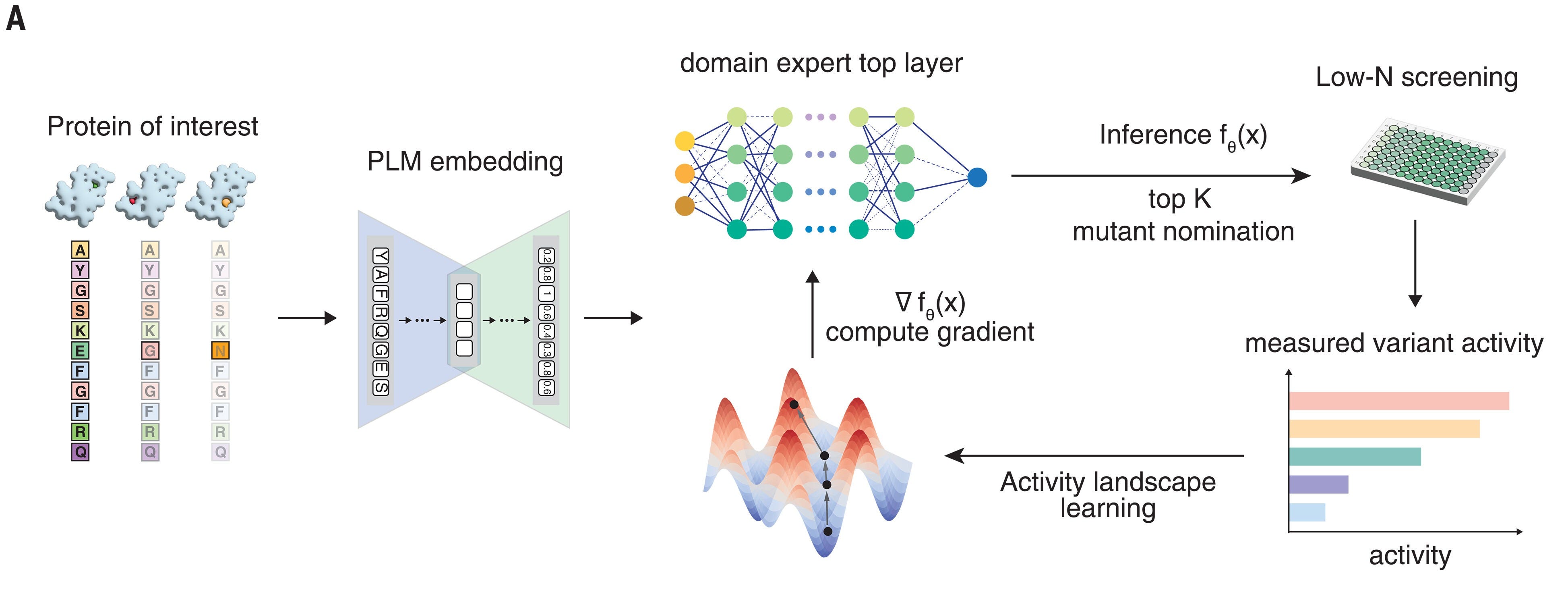
The model is built on top of ESM2, a foundation protein language model. ESM2 is a BERT-style masked language model: it takes a protein sequence and embeds each residue into a high-dimensional vector. It was trained on a large-scale dataset of protein sequences, UniProt. Once an embedding of a protein sequence is obtained, one can use it for different tasks, including prediction of structure (e.g. as in ESMFold); or one can average the embeddings to obtain a fixed-length digest, which can be used for the same kinds of things that you would use a sentence embedding language model for, e.g. RAG, recommendation, clustering, prediction, etc. On top of the average embedding, they put a prediction head, which is a simple random forest. They get initial data by synthesizing random variants; then, they engage in an active design process, using the model to predict the next mutation to try. The selection process is straightforward; they simulate all single residue changes, and pick the top N candidates that the model predicts.
If that sounds straightforward, it’s because it is: once you have a really strong base model, the ability to fit a basic regressor, and the ability to close the loop, you’re off to the races. Ultimately, all of this is made possible by the confluence of available tools, including the ability to read and write sequences at will; databases where people share their data; open models that can edited, re-mixed, fine-tuned and shared; and lab automation practices that make assays easier.
This translates into a step change in the speed of protein optimization, powered by a whole ecosystem of artifacts: foundation models, yes, but also datasets, databases, atlases, computational tools, and conventional models. As an aside, this new state of affairs has important applications in neuroscience, e.g. accelerating the design of new proteins for measuring and affecting the brain. So much of what neuroscience does is downstream of protein design: genetically encoded calcium and voltage indicators (GECIs, GEVIs); optogenetics; chemogenetics with DREADDs; barcoding, MAP-seq, BRIC-seq, Connectome-seq for mapping neural circuits. Heck, the biggest blockbuster brain drugs in decades are (modified) peptides: the GLP-1 agonists, which not only help regulate blood sugar and control body weight, but look promising to treat addiction and degenerative disorders like AD and PD.
Lessons to be learned
Let’s think about how these lessons from synbio would translate to the context of neuroscience. Are we there yet?
Where’s the data? Does it have sufficiently high entropy to have a wide range of validity? Where’s our PDB and UniProt? Is the data sufficiently well-annotated and cross-referenced, in a format that facilitates learning at scale?
What’s the coverage of our data? Do we have e.g. the equivalent of sequences from all the relevant organisms? A full genome from a single organism? Does it rise to the level of an atlas or a map, aspiring to have full coverage? A random sample? A non-random, non-representative sample?
What means do we have to actuate the system? What are our knobs (e.g. the equivalent of the ability to synthesize new proteins)? Are our observations and models matched to our knobs?
How are we validating our models? Do we have the means of taking our models and validating them in a closed loop, like we validate new protein designs?
On the data side, we have large-scale databases of neural activity like DANDI and OpenNeuro, as well as individual large-scale datasets like those from the Allen Institute, IBL, HCP, etc. The data can be substantial; around 10,000 hours or more from each of spikes, LFPs, sEEG, fMRI and EEG. This represents many PhDs worth of data collection, heroic efforts distributed across hundreds of laboratories. Clearly, these are a boon to the dry lab and the computational neuroscientist, and a starting point for foundation models.
But do they rise to the level of an atlas? It’s difficult to simultaneously obtain high physical coverage for recordings (i.e. covering the whole brain), spatial precision, and high coverage in task space. Even the highest dimensional recordings vastly undersample neural activity; if you add up all the spikes in the DANDI, that’s still–back of the envelope–less than the number of spikes you generate in your (human) brain every second. Furthermore, we’re getting, for the most part, relatively narrow slices of behavior, focusing on controlled and reproducible behaviors. Imagine building a DNA language model sampling from a single chromosome of a population of yeast cells; it’s not nothing, but it is a narrow slice of the space of possible genes, and a foundation model of the first yeast chromosome would have limited applicability outside of yeast genetics.
I’d argue that the more bio-centric atlases in neuroscience, e.g. cell type atlases, FlyWire, etc. tend to be more complete (i.e. have better coverage) than the neural activity atlases. But even in the best cases, we don’t always have the bridges that allow us to cross scales. FlyWire is wonderful as a complete connectome of the fly brain; but to translate that to simulated neural activity in a reliable way, we’d also want the transcriptomic background of each of the neurons, the distribution of receptors, and a complete characterization of the electrical activity of these neurons. This last one is an example of a bridge; the equivalent of PDB, something that takes us from one level (connectome → sequence) to another (electrical activity → protein folding). One of our highest priorities in the next 10 years of neuroscience should be to build the equivalent of PDB+UniProt for neuroscience in species that are phylogenetically close to humans; ideally, in humans themselves. That means genetic background; cell atlases and transcriptomes; mesoscale and microscale, molecularly-annotated connectomes; neural activity atlases; and calibration datasets (i.e. bridges) to stitch all these modalities into a coherent whole.
What about actuation? We’re certainly not at the level where we can synthesize neural activity the way we can synthesize proteins. The highest bandwidth and dimensionality means of actuation are at the sensory periphery (see, e.g. the retinal implants from Science, or even screens or headphones). Holographic optogenetics is still in its infancy, though I see a bright future for it. Focused ultrasound stimulation is rather low-dimensional, but it does have promise in precise modulation of neural activity in humans; and patterned stimulation across many sites in humans is still many years away. Finding new channels to precisely pattern stimulation is important; some of the recent work on biohybrid devices from Science and the next-gen neurotechnologies being funded by ARIA are moving us in the right direction.

Can we close the loop? I can think of a handful of cases where we have characterized, decoded, and optimized neural activity in a loop. One example, on the vision side, is that of inception loops, which have demonstrated finding maximizing stimuli for visual neurons (see refs 1, 2, 3, 4). A different example is nudging neural activity using holographic optogenetics (see refs 1, 2). These are currently heroic experiments, but they show a glimpse of what closed-loop design might look like. Foundation models, which are differentiable end-to-end, and can therefore be searched through gradient descent, have a clear role to play in closed-loop stimulation; but the means of stimulation and recording have to exist.
A call to action
All of that is a long-winded way of saying that our ambitions in neuroscience–solving all neurological disease, understanding intelligence and consciousness, etc.–are mismatched with our tools and datasets. Foundation models are one area of opportunity: leverage existing and future datasets to find good representations of neural data, make predictions, and optimize in a closed loop, leveraging the differentiability of deep learning models.
That cannot occur in a vacuum, however: the datasets that we collect, and the ecosystem of tools to read and write neural activity, cell identity, synapses, and connections should be matched in a virtuous circle. That will, almost assuredly, involve doing large-scale, hypothesis-free neuroscience focused on tooling and data, including in innovative structures like focused research organizations and coordinated research programs. Hypothesis-free science sometimes gets a bad reputation, but I think it can be most easily justified by epistemic humility: with billions of neurons, trillions of connections, thousands of cell types, hundreds of areas and receptors, maybe the first thing we should do is catalog these dang things, and then figure out how to poke at them to obtain causal models.
I can foresee, 10 years from now, many more neural atlases and databases, distilled into multiple application-specific foundation models and biophysically detailed models, cross-linked and validated through new neurotechnology. At the Amaranth Foundation, we philanthropically fund ambitious neuroscience projects led by ambitious mission-driven individuals; and my role is to lead our efforts in NeuroAI. Of course, I’m not a neutral party in all of this, but I think it’s a compelling vision for neuroscience.
I have hobbies.
Patrick, I really enjoyed this piece! I left a comment on LinkedIn about the need for a ‘Neural PDB,’ but one thing I keep thinking about is how this all plays into NeuroAI. Synthetic biology is already leveraging foundation models for designing and optimizing biological systems, but we don’t yet have that kind of closed-loop cycle in neuroscience.
What is the biggest bottleneck for bringing foundation models into NeuroAI? Is it more about better neural datasets, improving actuation (BCIs, optogenetics, etc.), or something else?